Chapter 8. Sex Determination and Sex Linkage
The Genetics of Sex Determination
Sex determination is an intricate series of physiological events and patterns that are often controlled through hormonal expression. Sexual characteristics can also be triggered by a change in hormonal balance, sometimes determined by an environmental change. In some organisms such as fruitflies sex is determined by a ratio of X chromosomes to the number of autosomal sets. A 1:1 ratio pro- duces a female, any deviation (1:2 etc.) produces a male.
For higher organisms, like humans, biological sex is determined by genes on the sex chromosomes. In humans, as in other animals, biological sex determination involves the XY system. The sex in which the sex chromosomes are not the same is referred to as heterogametic, or heteromorphic. Thus, in the case of humans, biological females are referred to as the homomorphic sex, and males are heteromorphic. Normal female karyotype contains two X chromosomes. Variations of the XX state include a missing X chromosome, XO (Turner Syndrome), as well as multiples of extra X chromosomes such as XXX and XXXX.
Normally, the XY karyotype is associated with a biological male individuals. Chromosome variations of the XY karyotype include extraX chromosome (Klinefelter syndrome, 47, XXY), as well as XXXY, XXXXY variations.
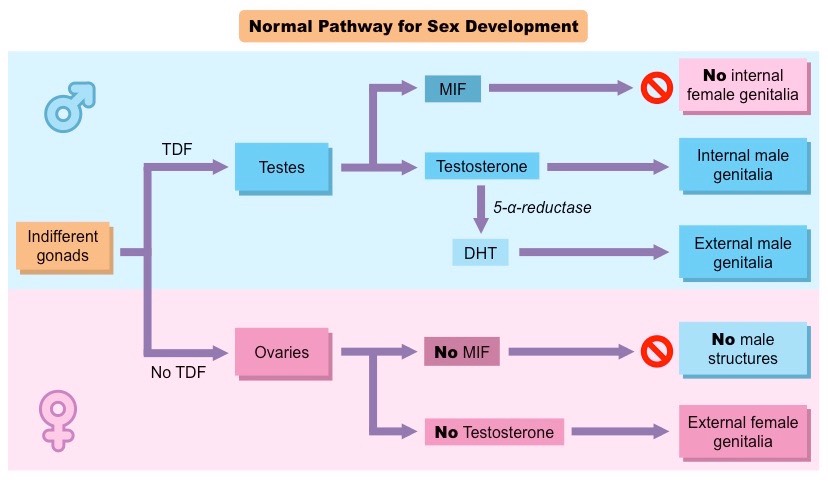
Every developing human embryo contains both male and female reproductive tracts up to the 6th week of development. By the 7th week of development, the SRY gene, if present (normally on the Y chromosome) and functioning, triggers testes development. Testes begin producing extra testosterone, compared to the baseline testosterone production by adrenocortical cells in both male and female embryos by the 8th week of gestation. If the fetus produces testosterone and the anti-Müllerian hormone (AMH) gene products from the Y chromosome, these molecules elicit cellular signaling events, such as producing Müllerian-inhibiting factor (MIF) that lead to the destruction of the female-specific Müllerian ducts.
A chromosome in a diploid organism is called hemizygous when only one copy is present. Hemizygosity is observed in the heterogametic sex when a gene is located on a sex chromosome.
Sex-linked inheritance
Sex linkage references genes located on sex chromosomes. Sex-linked inheritance is in contrast to the inheritance of traits on autosomal chromosomes, where both sexes have the same probability of inheritance. Since humans have many more genes on the X than the Y, there are many more X-linked traits than Y-linked traits.
X-Linked Recessive Disorders
X-linked recessive traits are expressed in all heterogametics, but are only expressed in those homogametics that are homozygous for the recessive allele. For example, an X-linked recessive allele in humans causes hemophilia.
Hemophilia is caused by a deficiency in blood clotting factors, which impairs the body’s ability to control blood clotting or coagulation – a necessary function to control bleeding if a blood vessel is ruptured. Hemophilia A (clotting factor VIII deficiency) is the most common form of the disorder, present in about 1 in 5,000–10,000 individuals with XY karyotype. Hemophilia B (factor IX deficiency) occurs in around 1 in about 20,000–34,000 XY births. A non- sex-linked hemophilia C due to coagulant factor XI deficiency, which can affect either XX or XY individuals, is more common in Jews of Ashkenazi (east European) descent but rare in other population groups.
Hemophilia is much more common in individuals with XY karyotype than those carrying XX karyotype, because XY individuals are hemizygous and therefore express the trait when they inherit one mutant allele. In contrast, an XX individual must inherit two mutant alleles, a less frequent event since the mutant allele is rare in the population. Tsarevich Alexei of Russia was the most famous sufferer of X-linked hemophilia, and his disease may have played an important role in the overthrow of the imperial regime in Russia in 1917, which changed the course of history for millions of people.
Other examples of X-linked recessive disorders include the Duchenne Muscular Dystrophy (progressive weakness and pseudohypertrophy due to replacement of muscle by fat and fiber) and Red-green color blindness (the genes for the red and green color receptors are located on the X chromosome).
X-Linked Dominant Disorders
An XX or XY child of a mother affected with an X-Linked dominant trait has a 50% chance of inheriting the mutation and thus being affected with the disorder. All XX children of an affected father will be affected (daughters possess their fathers’ X-chromosome). No XY children of an affected father will be affected (sons do not inherit their fathers’ X-chromosome).
Examples of X-linked dominant disorders include Incontinentia Pigmenti and vitamin D resistant rickets. Incontinentia Pigmenti is exclusively seen in XX individuals. It is presumed lethal in XY individuals. Characterized by swirling patterns of melanin, inflammatory skin lesions, missing teeth. The symptoms of vitamin D resistant rickets include growth failure, fractures that are very difficult to heal, spinal cord compression, hearing loss.
Mitochondrial Inheritance
Mitochondria contain circular DNA, thought to be derived from an ancient symbiosis with bacteria. All mitochondria are inherited from the mother. Thus, the oocyte contributes both genetic material in the nucleus and genetic material from the mitochondria.
Mitochondria are key to energy management for cells. Mutations in mitochondrial DNA lead to aberrations in energy management. Brain, eye, and muscle are high users of energy and so are the organs most often affected by mitochondrial disor- ders.
Sex Determination
In the animal world, environmental (rather than genetic) determination of sex is common.
Some organisms maintain both sexes in the same body.
By the 6th week of development in humans, both 46, XX and 46, XY embryos have the same sex organs.
By the 7th week of development, the SRY gene on the Y chromosome in 46, XY embryos produces testis-determining factor, TDF, that triggers testes development. TDF is a protein encoded by the SRY gene, normally on the Y chromosome. This protein is a transcription factor, which means it attaches (binds) to specific regions of DNA and helps control the activity of particular genes. TDF causes a fetus to develop as a biological male.
The presence of the SRY gene product activates other genes, found on the X chromosome and the autosomes, which further stimulate the development of male sexual organs.
At the same time, male-female assignment is not straightforward. Various mutations in key sex-determining genes produce a continous spectrum of phenotypic sex expression.
Gonadal determination during normal human development
True hermaphrodites are individuals with complete sets of male and female genitals.
Genetically, true hermaphrodites are either true 46, XX or sex chromosome mosaics (XXXY/XY, XX/XY etc.).
Sometimes, karyotypic males can be phenotypically females (and vise versa) due to a variety of genetic (and environmental) factors.
The androgen receptor (AR)
Complete Androgen Insensitivity Syndrome (CAIS)
The principal mammalian androgens are testosterone and its more potent metabolite, dihydrotestosterone (DHT). The androgen receptor (AR) is a large nuclear receptor protein of at least 910 amino acids. Each molecule consists of a portion which binds the androgen, a zinc finger portion that binds to DNA in steroid sensitive areas of nuclear chromatin, and an area that controls transcription.
Testosterone diffuses from the circulation into the cytoplasm of a target cell. Some is metabolized to estradiol, some reduced to DHT, and some remains as testosterone (T). Both T and DHT can bind and activate the androgen receptor, though DHT does so with more potent and prolonged effect. As DHT (or T) binds to the receptor, a portion of the protein is cleaved. The AR-DHT combination dimerizes by combining with a second AR-DHT, both are phosphorylated, and the entire complex moves into the cell nucleus and binds to androgen response elements on the promoter region of androgen-sensitive target genes. The transcription effect is amplified or inhibited by co-activators or co-repressors.
Although testosterone can be produced directly and indirectly from ovaries and adrenals later in life, the primary source of testosterone in early fetal life is the testes, and it plays a major role in human sexual differentiation. Before birth, testosterone induces the primary sex characteristics of biological males. At puberty, testosterone is primarily responsible for the secondary sex characteristics of males.
CAIS is a recessive sex-linked disorder (a form of male pseudo-hermaphroditism) affecting 1 in 20,000 XY individuals. CAIS is caused by a mutation in the Androgene Receptor gene (Xq11-12) that leads to lack of development of most of male physical characteristics and development of female genitalia. Individuals with CAIS have undescended testes that produce hormones inhibiting the development of uterus and fallopian tubes. The scientific and medical world accepts individuals with CAIS as females, though karyotypically they are males.
Gonadal Dysgenesis: Swyer Syndrome and True GD (46, XY and 46, XX Sex Reversal)
Gonadal dysgenesis is a reproductive system development disorder.
Swyer syndrome, or XY gonadal dysgenesis, is condition in which no functional testes are present in an externally female person whose karyotype is then found to be XY. Frequency: 1 in 30,000.
Swyer syndrome is a condition in which individuals with one X chromosome and one Y chromosome in each cell, the pattern normally found in males, have a female appearance. People with this disorder have female external genitalia and a normal uterus and Fallopian tubes. However, they do not have functional gonads (ovaries or testes). Instead, they have undeveloped clumps of tissue called streak gonads. These abnormal gonads often become cancerous, so they are usually removed surgically early in life. Some patients have no evidence of masculinization and have a female phenotype with the somatic signs of Turner syndrome. The presence of unilateral testis or a streak gonad and chromosomal mosaicism with both XO and XY cell lines (45,X/ 46, XY mosaicism) indicate the mixed gonadal condition.
SRY mutations that cause Swyer syndrome prevent production of the sex-determining region Y protein or result in the production of a nonfunctioning protein. A fetus whose cells do not produce functional sex-determining region Y protein will develop as a female despite having a Y chromosome.
People with Swyer Syndrome have streak gonads instead of testes (no germ cells or hormone-producing cells); they lack ovaries but have an otherwise normal female anatomy.
Mutations in the SRY gene have been identified in 15-20% of individuals with Swyer syndrome. Other genes involved:
- NR5A1 – autosomal (dominant mutation), hormonal production pathway;
- NR0B1 – X-linked, hormone pathway, endocrine function.
True sex reversal (complete gonadal dysgenesis) is found in individuals phenotypically displaying female secondary sex characteristics with 46, XY genotype and phenotypic males with 46, XX genotype (frequency 1 in 20,000 for each type). The origins of true GD are traced to an illegitimate crossing over in the SRY region during spermatogenesis or mutations of the SRY gene on the Y chromosome.
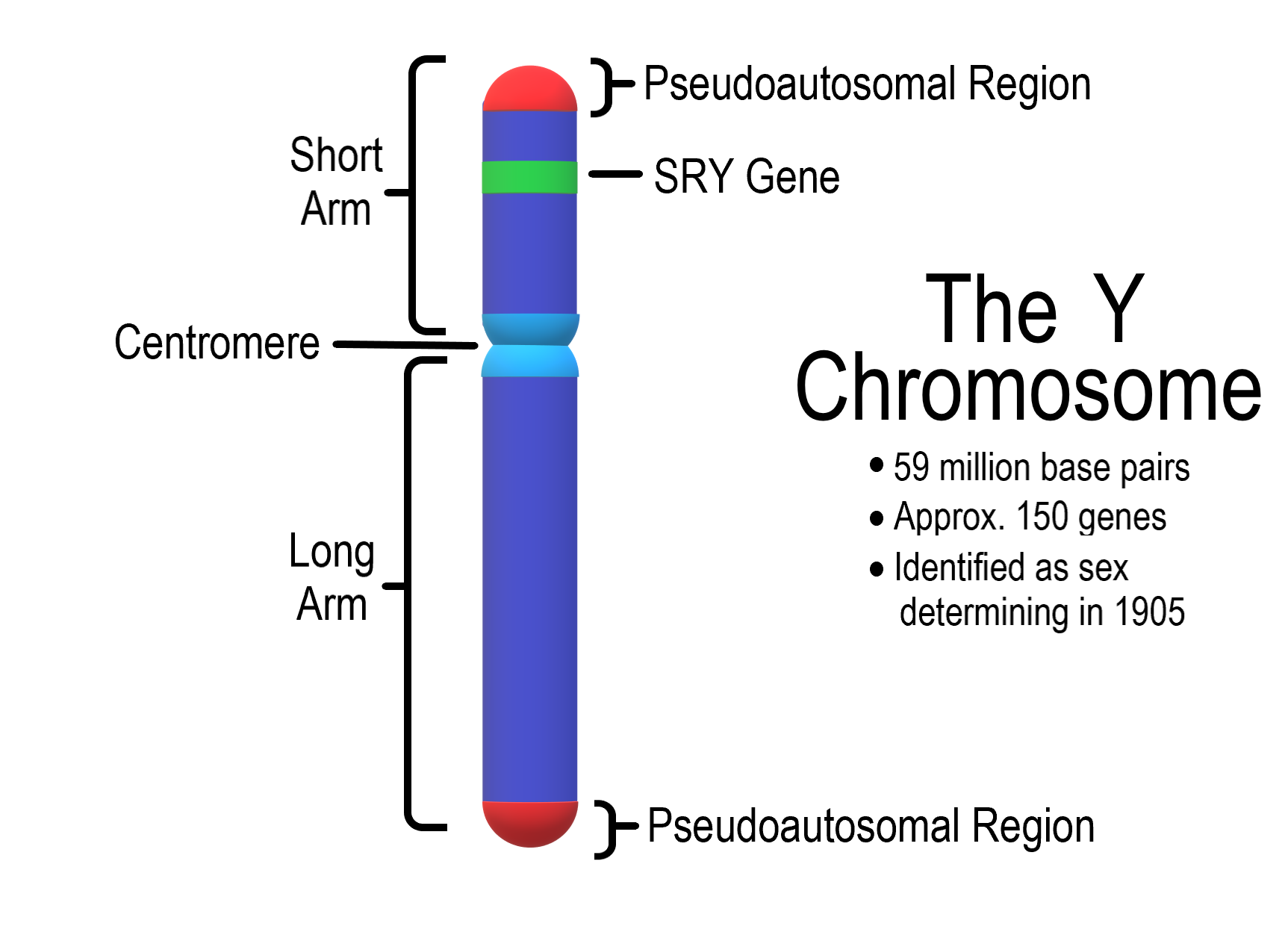
The Y Chromosome
The tips of both arms of the X and Y chromosomes are homologous (pseudoautosomal), which allows X and Y to pair during meiotic divisions of gametogenesis.
Pseudoautosomal regions of X and Y undergo normal crossing over in prophase I of spermatogenesis.
Rarely, an “illegitimate” crossing over occurs within the region just below the pseudoautosomal region, where the SRY gene is located.
The resulting Yp translocation leads to SRY loss in 46, XY fe- males and a gain of SRY in 46, XX females.
5-𝛼 (alpha) Reductase Deficiency
The enzyme 5-𝛼 reductase converts testosterone to dihydrotestosterone (DHT), which exerts an even stronger effect on the developing body than testosterone. T and DHT regulate the development of secondary sexual characteristics.
5-𝛼 reductase deficiency is an autosomal recessive condition. In this condition, the internal genitalia of males develop normally but the external genitalia resemble that of females.
The infant is typically raised as a girl but then at the next testosterone peak at puberty the external genitals suddenly become obviously male.
Key Takeaways
- Genes that regulate the development of human sexes are located on sex chromosomes.
- Functional alterations in sex-determining genes lead to ambiguous physiological sex characteristics.
A chromosome carrying genes that are involved in the determination of biological sex as well as the development of sexual characteristics in an organism.
A chromosome that is not a sex chromosome. There are 22 pairs of autosomes in humans.
